 新安潤コンサルティング Since 2009
新安潤コンサルティング Since 2009- Hot Line:
+(86)15801695345 - E-mail:
nar@china-reach.net
 中文
中文 English
English
中国薬品補助材料登録|新安潤コンサルティング株式会社
一、輸入、国内原料医薬品、医薬品補助材料、医薬品包装材料の登録
| 法的根拠 |
医薬品中華人民共和国法、医薬品中華人民共和国法施行規則、医薬品登録管理措置 原料医薬品、薬用賦形剤及び薬包材の審査及び承認に関する総局のお知らせ(2017年第146号) 医薬品関連審査・承認・監督作業の一層の充実に関する国家医薬品局のお知らせ(2019年第56号) |
| 適用範囲 | 国内中華人民共和国で開発、生産、輸入、使用される賦形剤。 |
| 登録情報の要件 | 附属書1は、生薬、医薬品賦形剤及び薬包材の審査及び承認に関する総局の公告の要件を満たすものとする |
| 登録の種類 |
輸入賦形剤の登録、更新、年次報告書。 国内補助材料の登録、更新、年次報告書。 |
| 受付機関 | 国家医薬品局医薬品レビューセンター(CDE) |
二、輸入および国産補助材料登録の目標
顧客の輸入または国内補助材料登録のための信頼性の高い登録コンサルティングサービスを提供し、製剤製品に関連するレビューを支援し、顧客製品が技術レビューに合格し、製剤企業が「輸入医薬品登録証明書」/「医薬品登録証明書」または医薬品承認番号を取得し、CDEが賦形剤登録番号の「技術レビュー」マーク、すなわち「A」ステータスを達成するのを助けます。
三、サービスの内容
| 1.輸入補助材料のための登録エージェントサービスを提供します。 | 4. 登録書類の審査、翻訳、完成、作成、提出 | 7. 賦形剤登録情報の技術移転。 |
| 2. 申告情報の提供に関する概要を作成する。 | 5. 検査申請、サンプル送信、追跡、問題解決の登録 | 8. 登録された賦形剤年次報告書の提出。 |
| 3. 登録情報のギャップ分析を行い、リスクを予測し、登録賦形剤が技術レビューに合格するための補正措置を提案する。 | 6. 補助材料登録は、完全な追跡、補足質問の回答、および補足応答情報の送信を通じて行います。 | 9. 重要な変更、中程度の変更、小さな変更、および基本情報の変更を含む、賦形剤登録情報の更新。 |
四、輸入、国産補助材料登録プロセス
| ①「原料薬、薬用補助材料及び薬包装材料申告登記表」に記入 | ②CDEに登記資料を提出 | ③登記資料の審査、システムより登記番号を公示、状態は”1”となる | ④製剤と関連の技術審査 | ⑤登記番号に標記を付け、状態は”A”となる |
五、中国における医薬品包装材の規制基準とモデル
中国薬局方は、国家の医薬品規格システムの重要な一部として、公布された「中華人民共和国薬物管理法」、「中華人民共和国薬物管理法実施条例」、ガイダンスとしての「医薬品包装材料登録データ要求事項」および各種技術指導書、さらに医薬品包装材料の国家規格(YBB規格と呼ぶ)と合わせて、医薬品包装材料の規制のための規制システムを形成している。 国家薬品監督管理局告示(2019年告示第56号)の内容によると、中国における医薬品包装材料の規制は、従来の登録管理制度から、医薬品製剤または原薬との関連技術審査を伴う登録制度に変更され、承認審査がより効率的かつ科学的に行われるようになりました。
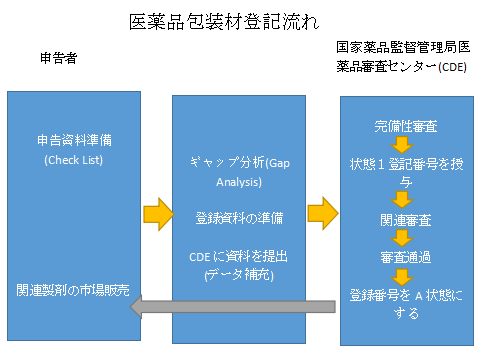
六、医薬品包装材の登録のためのステップ
– CDEポータル上で医薬品パッケージの基本情報を入力し、CD-ROMの形でCDEに登録データを提出する。
– CDEは、登録情報を受け取ってから5営業日以内に、登録情報が完全であるかどうかを確認します。
– CDEは登録情報を公開し、Iステータスの登録番号を付与します(登録番号の構成:B+4桁の年度番号+7桁のランニングナンバー)。
– 登録番号は、医薬品パッケージが関連する審査に合格した場合、Aステータスに有効化されます。
七、医薬品添加物の登録情報要件
医薬品協会の審査・承認・規制に関する事項の更なる改善に関する国家薬品監督管理局の発表」(2019年第56号)では、医薬品包装材を用途に応じて以下のように分類しています。
国内外の上場企業の医薬品に使用された実績がないもので、以下のものがあります。
○1.1 新規の分子構造を有する賦形剤及び1.2,1.3に該当しない賦形剤。
○1.2 使用実績のある賦形剤のうち、化学構造が単純に変化したもの(塩基、水和物など)をいいます。
○1.3 使用実績のある2種類以上の賦形剤を共重合して得られた賦形剤。
○1.4 使用実績はあるが、投与経路が変更になる添加物。
使用実績のある2つ以上の賦形剤を共同で処理して得られた賦形剤と
○2.1 中国薬局方/USP/EP/BP/JP に収載されていない賦形剤
○2.2 USP/EP/BP/JP のいずれかに収載されているが、国内で販売されている医薬品には使用されていない添加物。
○2.3 USP/EP/BP/JP のいずれかに収載されているが、中国薬局方には収載されていない添加物。
○2.4 中国薬局方に収載された賦形剤
食品や化粧品への使用実績があるものや
○3.1 口腔製剤に使用される食品安全に関する国家規格を有する賦形剤
○3.2 外用剤に使用される化粧品の国家規格または業界規格を有する賦形剤。
○その他
提案されている製剤の投与経路: ○注射 ○吸入 ○眼球 ○外用・舌下 ○経皮 ○口腔 ○その他
ソース: ○動物またはヒト ○鉱物 ○植物 ○化学合成 ○微生物による発酵またはバイオエンジニアリング ○その他
薬用補助材料登録資料表
| 資料項目 | 内容 | 1.1* | 1.2* | 1.3* | 1.4* | 2.1* | 2.2* | 2.3* | 2.4* | 3.1* | 3.2* |
| 1 | 登録者基本情報 | + | + | + | + | + | + | + | + | + | + |
| 2 | 補助材料の基本情報 | + | + | + | + | + | + | + | + | + | + |
| 3 | 3.1(1)工程概要 | + | + | + | + | + | + | + | + | + | + |
| 3.1(2)工程詳述 | + | ± | ± | ± | ± | ± | – | – | ± | ± | |
| 3.1(3)商業生産のバッチ原則、ロット範囲と根拠を説明する | + | + | + | + | + | + | + | + | + | + | |
| 3.1(4)設備 | + | + | + | + | + | + | + | + | + | + | |
| 3.2.1 重要な材料コントロール情報 | – | – | – | + | – | – | + | + | + | + | |
| 3.2.2 マテリアルコントロール情報詳細 | + | + | + | – | + | + | – | – | – | – | |
| 3.3 キーステップと中間体の制御 | + | + | + | + | + | + | – | – | + | + | |
| 3.4.1プロセス安定性評価 | – | – | – | + | + | + | + | + | + | + | |
| 3.4.2 プロセス検証 | + | + | + | – | – | – | – | – | – | – | |
| 3.5 生産プロセスの開発 | + | ± | ± | – | ± | ± | – | – | – | – | |
| 4 | 4.1.1(1)構造確認情報 | + | + | + | + | + | + | ± | – | + | + |
| 4.1.1(2)構造実証研究 | + | ± | + | – | – | – | – | – | – | – | |
| 4.1.2 理化性質 | + | ± | ± | ± | ± | ± | – | – | ± | ± | |
| 4.2.1 不純物情報 | + | + | + | + | + | + | + | + | + | + | |
| 4.2.2 不純物の研究 | + | ± | ± | ± | ± | ± | – | – | ± | ± | |
| 4.3.1 機能特性情報 | + | + | + | + | + | + | + | + | + | + | |
| 4.3.2 機能特性の研究 | + | + | + | ± | ± | ± | – | – | ± | ± | |
| 5 | 5.1 品質基準 | + | + | + | + | + | + | + | + | + | + |
| 5.2 分析方法の検証 | + | + | + | ± | + | + | – | – | ± | ± | |
| 5.3 品質基準制定根拠 | + | + | + | + | + | + | + | + | + | + | |
| 6 | バッチ検査報告書 | + | + | + | + | + | + | + | + | + | + |
| 7 | 7.1 安定性のまとめ | + | + | + | + | + | + | + | + | + | + |
| 7.2 安定性データ | + | + | + | + | + | + | + | + | + | + | |
| 7.3 補助材料の包装 | + | + | + | + | + | + | + | + | + | + | |
| 8 | 薬理毒理研究 | + | + | + | + | + | ± | ± | ± | ± | ± |
説明:
+関連資料を提供する必要がある項目
– 関連資料を提供する必要がない項目
±必要に応じて関連資料を提供する項目
備考:*
国内外の医薬品の中で使用歴史ないのは
1.1 新しい分子構造の補助材料及び第1.2、1.3に属さない補助材料。
1.2 既存の使用履歴による補助材料は簡単な化学構造により変更される(例えば、塩基、水和物など)。
1.3 両者及び両者以上の既存使用歴史の補助材料は共に処理して得られた補助材料である。
1.4 使用歴史がありますが、給薬の経路を変えた補助材料です。
国内外に上場する薬品の中に使用歴史があるもの
2.1 中国薬典/USP/EP/BP/JPは全部未収録の補助材料です。
2.2 USP/EP/BP/JP中の一つはすでに記載されていますが、国内で薬品に使用される補助材料が発売されていません。
2.3 USP/EP/BP/JP中の一つはすでに記載されていますが、中国薬局はまだ補助材料を受け取っていません。
2.4 中国薬局方は既に補助材料を受け取りました。
食品或は化粧品の中で使われている歴史があります。
3.1 食品安全国家基準を有する経口製剤の補助材料。
3.2 化粧品国家又は業界標準を有する外用製剤の補助材料。
注釈:
(1)高リスク薬用補助材料は、一般的に動物源または人間源の薬用補助材料を含みます。注射剤、眼用製剤、吸入剤などに使われる薬用補助剤です。高リスク補助材料の登録資料の要求については、補助材料の特定の製剤における応用及び相応の技術要求に基づき、必要に応じて提供することができます。または審査の過程で特定の製剤及び補助材料の製剤における応用状況に基づき、必要に応じて資料を補充します。
(2)既に使用されている使用履歴の補助材料に対して、当該補助材料が供給経路の歴史最大使用量を超えた場合、関連安全性データなどの資料を提供しなければならない。
(3)プレミックス補助材料については、製剤における応用及び調合組成における各補助材料の成分状況に基づき、適切な資料を選択して登録しなければならない。
(4)以上の登録資料の分類要求は登録者資料として準備された指導であり、薬品審査センターは製剤の技術審査に基づいて資料補充要求を提出することができる。
(5)補助材料の分類によって、登録資料は3.2.1と3.2.2、3.4.1と3.4.2、4.1.1(1)と(2)の中で研究資料のセットを提供すればいいです。
医療機械登録 CFDA:「医療機器監督管理条例」及びCFDA第4号、7号などの法令により、予定発売販売、使用する医療機器の安全性、有効性をワンストップ式の技術指導を提供し、CFDA審査を通過させ、円滑に発売、使用できるように支援します。
国は、医療機器をリスクの程度に応じて分類し、管理します(*特定の分類は、医療機器の分類に関する規則(国家食品医薬品局令第15号)を参照することができます)
第1のカテゴリーは、リスクの低さであり、定期的な管理は、その安全で効果的な医療機器を保証することができます。
第2のカテゴリーは、中リスクであり、安全で効果的な医療機器を確保するために厳格な管理が必要です。
第3のカテゴリーは、リスクが高く、安全で効果的な医療機器を確保するために、管理を厳格に管理するための特別な措置が必要です。
一、製品
- 医療機器製品は、ファイリング/登録の分類管理システムを採用しています。 医療機器製品の第1分類は、製品のファイリングに適用され、第2および第3のカテゴリは、製品登録に適用されます。
- 国内生産製品
医療機器製品の第1のカテゴリーのファイリングは、記録者が、その地域に所在する地方自治体の食品医薬品監督管理部門に記録資料を提出し、その情報が要件を満たしていることを前提に、通常、処理時間制限が5営業日を超えることなく行われるものとする(地域管理部門間の差異)。
医療機器製品の第2のカテゴリーの登録は、登録者が、中央政府直轄の地方、自治区、または地方自治体の食品医薬品監督管理部門に登録申請書を提出しなければならない。 受領部門は、技術レビューのコメントを受け取った後、20営業日以内に承認決定を行います。
医療機器製品の第3のカテゴリーの登録は、登録者が国務院の食品医薬品監督管理部門に登録申請書を提出しなければならない。 受領部門は、技術レビューのコメントを受け取った後、20営業日以内に承認決定を行います。
- 海外からの輸入品
輸入品のファイリング/登録は、国務院の食品医薬品監督管理部門に提出する必要があり、対応するファイリング/登録資料に加えて、医療機器の市場販売を許可するホスト国(地域)の所轄官庁の証明書を提出し、我が国の領土に設立された代表機関または指定国内企業法人を代理人として提出する必要があります。
| 二、生産 | 三、経営 |
| 医療機器製造の第一級は、記録管理に適用されます:生産企業は、その要件を満たす証明書を提出し、提出するために、市レベルの人民政府の食品医薬品監督管理部門に提出します。 | 医療機器の最初のカテゴリは、ファイリングやライセンスを必要としません |
| 第2、第3の医療機器製造は、生産ライセンス管理に適用されます:生産企業は、中央政府直轄の地方、自治区、または地方自治体の食品医薬品監督管理部門に生産許可を申請し、その要件を満たす証明書を提出する必要があります。 審査の対象となる医療機器の製造許可は、5年間有効です。 | 医療機器事業の第2のカテゴリーは、記録管理に適用されます:事業会社は、その要件を満たす証明書を提出し、提出するために、市レベルの人民政府の食品医薬品監督管理部門に提出します。 |
| 医療機器製造の第三のカテゴリーは、ライセンス管理に適用されます:事業会社は、その要件を満たす証明書を提出するために、地方自治体の食品医薬品監督管理部門にライセンスを申請する必要があります。 審査の対象となる医療機器の営業許可は、5年間有効です。 |
八、弊社のサービス
– 医薬品パッケージの登録に関する相談
– 医薬品の登録
– 登録書類の翻訳
– 登録内容の見直しに関するフォローアップ
– 医薬品原料に関する年次報告
– ラボのコミッショニングとテストの監督
– 公式問い合わせの連絡
GMP認定|新安潤コンサルティング株式会社
![]() GMP 準拠および認定
GMP 準拠および認定
 |
システム構築、完成、監査、模擬検査から公式認証検査まで、一括サービスを提供します 含まれるもの: |
| (1)中国GMP認証 | 国内企業GMP認証 | 海外企業のNMPAオンサイト検査 |
| (2)国際GMP認証 | 米国FDA、EU、オーストラリア、WHO、PIC/S GMP認証 | GMP 準拠/483/警告メッセージ修正 |
| (3)GMPプロフェッショナル翻訳 | GMPは、中国語と英語の通訳/翻訳をチェックします | GMPは、韓国、中国、日本の通訳/翻訳をチェックします |
| (4)GMPプロジェクト | 新しい/改装された工場(ワークショップ)の計画、設計、GMPコンプライアンスコンサルティング | |
| (5)R&Dラボシステム規制に準拠しています | 研究開発体制の構築・欠陥改善指導・監査等 |
![]() 検証測定コンサルティング
検証測定コンサルティング
(1)機器・機器確認サービス (2)プロセス/方法/システム検証サービス (3)コンピュータ化されたシステム検証サービス
![]() 国内登録
国内登録
| (1)輸入/国産医薬品の登録 | (2)国産医薬品の研究開発指導サービス | (3)輸入/国産健康食品登録 | (4)輸入/国産化粧品登録 |
|
|
||
|
|
||
|
|||
|
![]() 国際登録
国際登録
(1)医薬品類
|
|
|
|
|
|
|
|
|
(2)健康食品類
米国ダイエットサプリメント認証と新成分(NDI)は、米国食品添加物認証を通知します
EU食品サプリメント/添加物認証 EU新食品(Novel Food)および成分認証
医薬品上場許可書保有者(MAH)のホスティングサービス
薬政顧問
企業の年間薬政規制コンサルタント
医薬品の技術移転
登録者制度の規制を各地にプッシュし、常に政策情報を先取りする
登録者モデル計画、バインドR&D – 製造の両側の2つのシステム
受託者システム評価テンプレートは、登録者が適切なメーカーを見つけるのに役立ちます
登録者品質システムおよびリスクシステムは、製品登録のための最も信頼性の高いサポートを提供します
GMPシミュレーションチェックは、登録者が検証に合格し、保険に加入します
サプライヤーは、受託メーカーから製品テスト、原材料の選択まで、推奨されています
GMP/生産品質システムサービス
医薬品製造企業のGMPシステムの構築と実装:リスク管理、偏差と変更管理、データ信頼性、OOS/OOT調査、製品品質レビューなど
医療機器メーカーGMP+ISO13485システムの構築と実装
医薬品研究開発企業GLP+GMPシステムの構築と実施
栄養補助食品メーカーISO9000+BRC+GMPシステムの構築と実装
浄化プラント設計監査
GMPエンジニアリングプロジェクト設計レビュー
GMPエンジニアリングプロジェクト管理とサービス
GMPプラント監査および監査プロジェクト管理
品質監査と工場デューデリジェンス
海外現場シミュレーション品質監査(Mock Audit)
FDA/EDQMのGMP現状評価
第三者登録審査/GMPシミュレーションオンサイト検査、品質監査を提供します
製薬工場のM&AまたはMAHを目的とした第三者デューデリジェンスを提供する
プラントの製造レベル、能力能力、生産・運用管理能力、製品転送能力を評価します
登録者システム構築/MAH
当社のサービス
- GMP遵守と認証
- 医薬品添加物、医薬品包装材料、APIの登録サービス
- 医薬品製造販売承認者(MAH)のホスティングサービス
- GMP/製造品質システムサービス
- クリーンルーム設計監査
- 品質監査と工場デューディリジェンス
サービスホットライン:(+81)090-6677-6889
北京本部:中国北京市朝陽区東三環北路甲26号 博瑞ビル A 座4階 D10
Tel: +86-10-6471-0683
上海オフィス:中国上海市閔行区浦星公路800号 中意国際広場D308室
Tel: +86-21-3478-3993
広州オフィス:中国広州市番禺区興南ビル445号 合誠ビル6階612室
Tel: +86-400-660-1329
メール:nar@china-reach.net

「化学品法规前沿」
