 新安潤コンサルティング Since 2009
新安潤コンサルティング Since 2009- Hot Line:
+(86)15801695345 - E-mail:
nar@china-reach.net
 中文
中文 English
English
- 医療機器規制制度の紹介
医療機器規制制度は、一連の法律、規制、基準、ガイドラインで構成され、これらが一体となって、研究開発、製造、販売、使用の全過程における医療機器の規制の基礎となっている。 医療機器規制制度は主に以下のレベルの規制で構成されている:
- 「医療機器監督管理条例」
医疗器械监管的基本法规,由国务院发布,涵盖了医疗器械的分类、注册、生产、经营、使用、监督管理和法律责任等方面,为医疗器械的监管提供了法律依据 国務院が発布した医療機器の監督管理の基本法規は、医療器材の分類、登録、生産、経営、使用、監督管理、法的責任などをカバーしており、医療器材規制の法的根拠となっている。
- 「医療機器登録申請管理弁法」
「医療機器登録申請管理弁法」は、医療機器の登録申請プロセスについて詳細に規定しており、登録区分、申請要件、技術審査、臨床試験要件、優先承認手続き、登録変更、登録継続などの内容が含まれる。
- 国家薬品監督管理局の公告とガイドライン:
医療機器のマスターファイル登録に関する公告、臨床試験ガイドライン、技術指導原則などを含み、これらの公告とガイドラインは医療機器の研究開発、登録と販売に具体的な操作指導を提供した。
- 国や業界の基準:
医療機器製品は、製品の安全性、有効性、品質管理などに関連する国家規格や業界規格に準拠する必要がある。例えば、GB/T 16886シリーズは医療機器の生物学的評価に関する国家標準である。
- 「医療機器生産品質管理規範」(GMP)
医療機器の製造管理及び品質管理に関する基準(GMP)は、医療機器の製造において、製品の品質の安定性及び信頼性を確保するために必要な品質管理事項を定めたものである。
- 「医療機器経営品質管理規範」(GSP)
医療機器の調達、検収、保管、輸送、販売など、医療機器業務における品質管理に関する要求事項を定めたもの。
- 医療機器有害事象モニタリングと再評価管理弁法
医療機器有害事象モニタリング及び再評価管理弁法は、医療機器の市販後有害事象のモニタリング、報告、分析及び処理の手順、並びに再評価の要件を規定している。
- 医療機器製品の監督管理
医療機器の監督管理は、リスク管理、全過程管理、科学監督、社会共同管理の原則に従っている。 国家は医療機器の分類管理を実施し、リスクの程度によって、第一分類は低リスク、第二分類は中リスク、第三分類は高リスクの3つに分類している。 それぞれの分類には、対応する管理要件と登録・申請プロセスがある。
- 第一類医療機器:製品届出管理を実行する
- 第二類、三類医療機器:製品登録管理を実行する
監督管理部門は下表の通り
| 分類 | 第一類製品 | 第二類製品 | 第三類製品 | |
| 登録 | 国産 | 市(市場局)届出 | 省(薬監局)登録 | 国家(薬監局)登録 |
| 輸入 | 国家(薬監局)登録(一類届出) | |||
| 生産 | 市(市場局)届出 | 省(薬監局)許可 | ||
| 経営 | — | 市(市場局)届出 | 市(市場局)許可 | |
- 医療機器製品の届出/登録プロセス
国家薬品監督管理局は、医療機器の安全性、有効性、品質を確保するために、医療機器製品の申請・登録プロセスを管理し、一連の手続きと要件を満たすことができる。
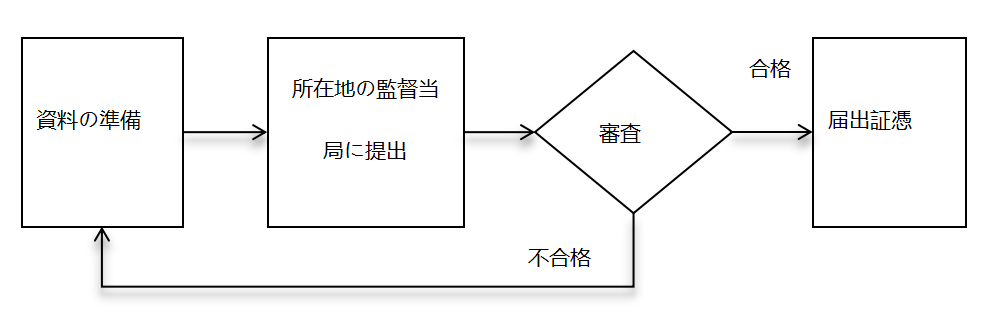
① 中国における第一類医療機器届出
- 届出情報の提出:届出人は、届出情報を当該地区の薬物監督管理を担当する市の担当部署に提出する。
- 出願番号の取得:監督管理部門は提出された届出資料を保存し、届出番号を付与する。
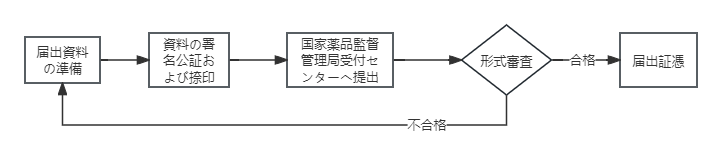
② 輸入第一類医療機器届出
- 届出資料の提出:届出人は国家薬品監督管理局に届出資料を提出する。
- 届出番号の取得:国家薬品監督管理局は提出された届出資料を保存し、届出番号を付与する。
③ 中国国内第二類、第三類医療機器登録
- 登録申請の提出:申請者は省、自治区、市直轄の薬品監督管理局または国家薬品監督管理局に登録申請書を提出する。
- 審査:関連部門が申請資料を審査する。
- 承認:審査合格後、医療機器登録証が発行される。
④ 輸入第二類、第三類医療機器登録
- 登録申請の提出:申請者は国家薬品監督管理局に登録申請を提出する。
- 審査:国家薬品監督管理局は申請書類を審査する。
- 承認:審査通過後、医療機器登録証を発行する。
二類/三類医療機器製品登録プロセス

- サービス内容
- 国内医療機器製品届出/登録サービス

- 海外医療機器製品届出・登録サービス
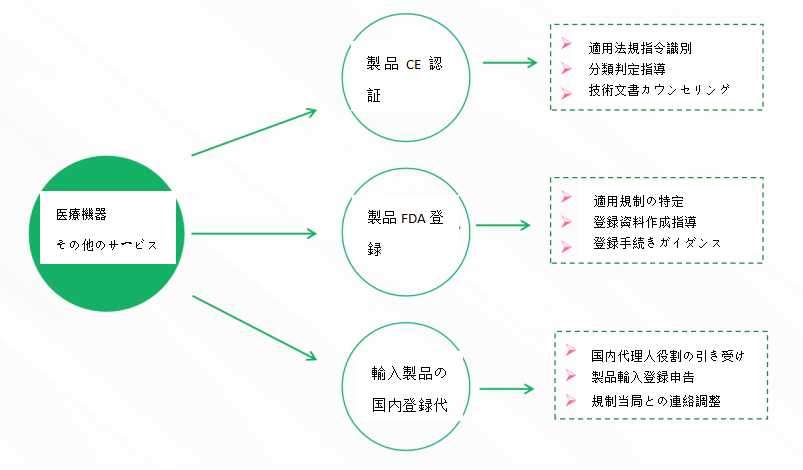
- 医療機器臨床評価サービス
| 医療機器臨床評価サービス | 臨床試験指導サービス | 治験実施計画書の作成・評価 |
| 治験実施医療機関の調査・審査 | ||
| 治験プロジェクト開始(プロジェクト申請、倫理審査、治験申請、契約締結、開始会議) | ||
| 治験遺伝子事務局承認申請コンサルティング業務 | ||
| 治験モニタリング業務 | ||
| 臨床試験品質監査 | ||
| 治験データ管理 | ||
| 臨床試験統計分析 | ||
| 臨床試験報告書作成、レビュー、修正 | ||
| 同種比較臨床評価 | 臨床評価コンサルティング(同種製品比較) | |
| 免除臨床比較説明(免除臨床試験カタログの製品について) |
サービスホットライン:+(86)-400-660-1329
北京本部:中国北京市朝陽区東三環北路甲26号 博瑞ビル A 座4階 D10
Tel: +86-10-6471-0683
上海オフィス:中国上海市閔行区浦星公路800号 中意国際広場D308室
Tel: +86-21-3478-3993
広州オフィス:中国広州市番禺区興南ビル445号 合誠ビル6階612室
Tel: +86-400-660-1329
メール:nar@china-reach.net

「化学品法规前沿」
